Le tadalafil possède une affinité marquée pour la PDE5, mais épargne en grande partie les isoformes PDE1, PDE2 et PDE11, réduisant ainsi le risque d’effets extra-caverneux. L’action se traduit par une augmentation contrôlée de la circulation sanguine locale, indépendante des variations alimentaires. Sa pharmacocinétique repose sur une absorption digestive rapide, un métabolisme hépatique par CYP3A4 et une distribution tissulaire large. La biodisponibilité reste stable, et l’équilibre plasmatique est atteint en quelques jours lors d’administrations répétées. Les interactions cliniquement significatives surviennent avec les inhibiteurs puissants de CYP3A4 tels que le kétoconazole. Dans la littérature pharmacologique, acheter cialis 20 mg est souvent associé à des schémas d’utilisation basés sur la durée prolongée de son action.
Structure-function relations of human hemoglobins
Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
Note: Performing your original search, "hemoglobin koln" symptoms, in PubMed Central will retrieve 8 citations.
Journal List > Proc (B ayl U niv M ed Cent) > v.19(3); Jul 2006
Proc (Bayl Univ Med Cent). 2006 July; 19(3): 239–245.
Copyright 2006, Baylor University Medical Center
Structure-function relations of human hemoglobins Related material:
From the Department of Pathology, Baylor University Medical Center, Dallas, Texas. PubMed articles by: Corresponding author: Alain J. Marengo-Rowe, MD, Department of Pathology, Baylor University Medical Center, 3500 Gaston Avenue, Dallas, Texas 75246. Abstract
In 1949 Pauling and his associates showed that sickle cell hemoglobin (HbS) belonged to an
abnormal molecular species. In 1958 Ingram, who used a two-dimensional system of electrophoresis
and chromatography to break down the hemoglobin molecule into a mixture of smaller peptides,
defined the molecular defect in HbS by showing that it differed from normal adult hemoglobin by
only a single peptide. Since then, more than 200 variant and abnormal hemoglobins have been
described. Furthermore, the construction of an atomic model of the hemoglobin molecule based on a
high-resolution x-ray analysis by Dr. Max Perutz at Cambridge has permitted the study of the
stereochemical part played by the amino acid residues, which were replaced, deleted, or added to in
each of the hemoglobin variants. Some of the variants have been associated with clinical conditions.
The demonstration of a molecular basis for a disease was a significant turning point in medicine. A
new engineered hemoglobin derived from crocodile blood, with markedly reduced oxygen affinity
and increased oxygen delivery to the tissues, points the way for future advances in medicine.
POSSIBLE CLINICAL DEVELOPMENTS IN HEMOGLOBIN
Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
Hemoglobin has played a spectacular role in the history of biology, chemistry, and medicine. This paper, written primarily for the clinician, is a brief outline of the complex problems associated with abnormal hemoglobins. The thalassemias have been intentionally omitted and will be presented in a separate publication.
Hemoglobin is a two-way respiratory carrier, transporting oxygen from the lungs to the tissues andfacilitating the return transport of carbon dioxide. In the arterial circulation, hemoglobin has a highaffinity for oxygen and a low affinity for carbon dioxide, organic phosphates, and hydrogen andchloride ions. In the venous circulation, these relative affinities are reversed. To stress theseremarkable properties, Jacques Monod conferred on hemoglobin the title of “honorary enzyme.” Ifwe call heme its active site, oxygen its substrate, and hydrogen ions its inhibitors, then hemoglobinmimics the properties of an enzyme. Therefore, it became evident that unraveling the properties ofhemoglobin was necessary to understanding the mechanism of hemoglobin function as it pertains torespiratory physiology.
In 1937, Dr. G. S. Adair gave Dr. Max Perutz crystals of horse hemoglobin (personal communication, Max Perutz, 1966). This started Dr. Perutz on the path that led to the elucidation of the structure of hemoglobin (1). For this endeavor he was awarded the Nobel Prize in chemistry in 1962.
In 1957 Ingram demonstrated that sickle cell anemia was caused by the replacement of one of the 287 amino acid residues in the half molecule of hemoglobin (2). This finding facilitated understanding of disease at the molecular level, since for the first time a point mutation in a structural gene was shown to cause the substitution of one amino acid in the protein controlled by that gene. Furthermore, the accumulation of the sickle cell gene in malarial regions of the world became a convincing illustration of evolution by natural selection (3). Persons with the sickle cell trait (HbA/S) have a selective advantage over normal individuals when they contract falciparum malaria because the parasite count remains low and lethal cerebral malaria is avoided.
To date, well over 200 hemoglobin variants have been described. The term “variant” rather than“abnormal” is preferred because most hemoglobins are not associated with disease. The lateProfessor Herman Lehmann at Cambridge University in England and his “musketeers” in differentparts of the world have been responsible for discovering many of these variants. Furthermore, asknowledge accumulated, it became evident that the structure-function relations of varioushemoglobins in stereochemical terms could be related to clinical symptomatology (4, 5). STRUCTURE OF HEMOGLOBIN
Hemoglobin comprises four subunits, each having one polypeptide chain and one heme group
(Figure 1). All hemoglobins carry the same prosthetic heme group iron protoporphyrin IX
associated with a polypeptide chain of 141 (alpha) and 146 (beta) amino acid residues. The ferrous
ion of the heme is linked to the N of a histidine. The porphyrin ring is wedged into its pocket by a
phenylalanine of its polypeptide chain. The polypeptide chains of adult hemoglobin themselves are
of two kinds, known as alpha and beta chains, similar in length but differing in amino acid
sequence. The alpha chain of all human hemoglobins, embryonic and adult, is the same. The
non-alpha chains include the beta chain of normal adult hemoglobin (α
fetal hemoglobin (α2 2), and the delta chain of HbA2. In some variants, the gamma genes are
duplicated, giving rise to two kinds of gamma chains.
Oxygen binds reversibly to the ferrous iron atom in each heme group. The heme group that has
become oxygen bound varies with the partial pressure of oxygen. The sigmoid shape of the oxygen
equilibrium curve shows that there is cooperative interaction between oxygen binding sites. Hence,
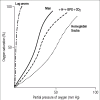
as oxygenation proceeds, combination with further molecules of oxygen is made easier. The oxygen equilibrium (or dissociation) curve is not linear but S-shaped and varies according to environments and species (Figure 2). At a partial pressure of oxygen of 100 mm Hg, the hemoglobin in the red cell is fully saturated with oxygen. The dissociation curve is plotted as percentage of oxygen saturation against partial pressure.
Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
The structure of hemoglobin has been extensively studied by x-ray analysis (6). The arrangement ofthe subunits—which is known as the quaternary structure—differs in the oxy- anddeoxyhemoglobin.
In human hemoglobin, the fit between the polypeptide chain is critical because the gap between two of the polypeptide chains in the hemoglobin molecule becomes narrower when oxygen molecules become attached to the ferrous atoms. This has been likened by Max Perutz to a molecular form of paradoxical breathing: unlike the lungs, the hemoglobin molecule contracts when oxygen enters and expands when oxygen leaves.
Compounds other than oxygen, such as nitric oxide and carbon monoxide, also are able to combine with the ferrous atom of hemoglobin. Carbon monoxide attaches itself more firmly to the ferrous atom than oxygen does. Once carboxyhemoglobin is formed, oxygen cannot displace carbon monoxide to any extent. This forms the molecular basis of coal gas poisoning.
In the body, the adequacy of the oxygen transport system depends on the adequacy of oxygenation of blood in the lungs, the rate and distribution of blood flow, the oxygen-carrying capacity of the blood (hemoglobin concentration), and the affinity of hemoglobin for oxygen so as to allow unloading of oxygen in peripheral capillaries. Hence, the availability of oxygen to the body may be altered by abnormalities at any point in this physiological pathway. In this paper, only the role of hemoglobin affinity for oxygen will be considered as variant forms of hemoglobin are discussed. SICKLE CELL HEMOGLOBIN
Sickle cell hemoglobin (HbS) has existed in humans for thousands of years. Dr. Konotey-Ahulu, a
Ghanaian physician, reports that among West African tribes, specific names were assigned to
clinical syndromes identifiable as sickle cell anemia (7). However, sickle cells were first described
in the peripheral blood of an anemic patient from the West Indies by the Chicago physician Robert
Herrick in 1910(8). While homozygous sickle cell anemia is the most common and severe form of
sickle cell disease (SCD), other sickling disorders combining HbS with beta or alpha thalassemia,
hemoglobin C, hemoglobin D, and other hemoglobins share a similar pathophysiology with
common as well as distinguishing clinical features.
HbS results from a single base-pair mutation in the gene for the beta-globin chain of adult
hemoglobin. An adenine-to-thymine substitution in the sixth codon replaces glutamic acid with
valine in the sixth amino acid position of the beta-globin chain (9, 10). This substitution yields the
electrophoretically distinct hemoglobin described by Linus Pauling in 1949 (11). In the
deoxygenated form of HbS, the beta-6 valine becomes buried in a hydrophobic pocket on an
adjacent beta-globin chain, joining the molecules together to form insoluble polymers (9). In sufficient concentration, these insoluble polymers give rise to the classical sickle morphology. This process causes severe damage to the red cell membrane. Sickled red cells may then aggregate and go on to cause microvascular obstruction. Also, these abnormal red cells adhere to endothelial cells (12) and can interact with various cytokines (13).
The process of microthrombosis and microembolization is the foundation of SCD pathology. Occlusion of the microvasculature by sickled erythrocytes causes painful crises, priapism, pulmonary emboli, and osteonecrosis, and ultimately damages every organ system including the retinae, spleen, liver, and kidneys. Many patients with SCD have hematocrits of 20% to 35% and chronic reticulocytosis. Clinical symptoms can be precipitated by fever, infection, excessive exercise, temperature changes, hypoxia, and hypertonic solutions. The clinical severity of the symptoms experienced is related to the concentration of HbS in the red blood cell and expression of other hemoglobins, endothelial factors, nitric oxide and other factors. Also, patients with SCD have a higher proportion of dense, dehydrated erythrocytes (14).
In about 11% of SCD patients under 20 years of age, stroke occurs because of stenotic cranial artery lesions, demonstrable by transcranial Doppler ultrasonography. A regular program of transfusion aimed at reducing the sickle cell population to <50% prevents about 90% of stroke cases. Unfortunately, the high risk of stroke returns after transfusion is discontinued (15).
Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
The surface of HbS consists mainly of hydrophilic amino acid side chains together with some smaller hydrophobic side chains. Since adult hemoglobin is present at a very high concentration within the red cell and yet appears to remain free from aggregation at all levels of saturation with oxygen, the amino acids on the surface of the molecule must be arranged so as to avoid attraction between adjacent molecules. Of the majority of hemoglobin variants with surface amino acid substitutions, only a minority are associated with any significant clinical abnormalities. Except for HbS, none of those more common hemoglobins found in the homozygous state, such as hemoglobins C, D, and E, are associated with any greater abnormality than mild anemia. The surface of hemoglobin A is therefore able to accommodate a variety of different amino acid changes without its structure or function being affected (16).
The valine-for-glutamic acid substitution has very little effect on the oxygenated form of HbS (17). However, when the concentration of deoxygenated HbS becomes sufficiently great, its properties differ markedly from those of deoxygenated hemoglobin A, causing the formation of insoluble fibers and bundles, which distort the red blood cell into the sickle shape.
Since the discovery of HbS, the clinical symptomatology and associated pathophysiology of SCD have gradually been elucidated (18). SCD is characterized by anemia and four types of crises: painful (vasoocclusive), sequestrative, hemolytic, and aplastic. Damage to the red blood cell membrane gives rise to reduced cell survival and chronic hemolytic anemia. If severe enough, this damage increases the risk of bilirubin gallstone formation, stroke, and heart failure. Also, the anemia is aggravated by the mechanical impedance to blood flow caused by sickled red blood cells, resulting in widespread vasoocclusive complications. Interestingly, the anemia to some degree can be protective against vasoocclusive complications, as it moderates the increase in viscosity associated with sickling in the microcirculation. Hence, judicious exchange transfusion therapy and blood transfusion is indicated for the prevention of pain crises, stroke, pulmonary hypertension, and other related conditions (19).
Blood transfusion not only increases the oxygen-carrying capacity of blood but also decreases thepercentage of cells capable of sickling. It is recommended that transfusion should be carried outwith phenotypically matched, leuko-reduced, sickle-cell–negative blood in order to attain aposttransfusion hematocrit of about 36%. (20). The complications of transfusion are well known and include allo- and autoimmunization, iron overload, and the transmission of infectious diseases such as hepatitis and HIV. Also, a considerable number of patients with sickle cell anemia worldwide have undergone successful bone marrow transplantation (21). Only selected patients are eligible for the procedure. Even then, bone marrow transplantation was associated with a 5% to 10% mortality, mostly from graft-versus-host disease.
Another approach to reducing the effect of HbS polymer formation has been to augment the production of fetal hemoglobin (HbF). Through population and clinical observation, it has long been recognized that higher blood HbF levels correlate with fewer clinical manifestations of SCD. Pharmacologic manipulation of HbF in the therapy of sickling disorders has been proposed since the mid-1950s. To date, several agents have been tried, but the safest and most effective has proven to be hydroxyurea(22). The mechanism of increased HbF production by hydroxyurea is not fully understood. Also, recent studies have found that hydroxyurea contributes to the production of nitric acid, a potent endothelial relaxing factor (23).
Numerous inflammatory markers associated with endothelial surfaces and white blood cells are elevated in SCD, including C-reactive protein. Baseline granulocyte counts are often increased. Leukocytosis itself is a risk factor for increased mortality (24). Finally, laminin, a constituent of the endothelial matrix that binds to the Lutheran antigen on red cells, is expressed on sickled red blood cells in greater quantities than on normal red blood cells (25).
Almost every aspect of hemostasis tending to hypercoagulability has been described in SCD (26). However, it is not known whether the hypercoagulability is the cause or the result of vasoocclusion. Thrombocytosis is due to hyposplenia, and platelet aggregation is increased (27). Antiphospholipid antibodies may be elevated, and protein C and S levels are decreased (28). Also, high levels of von Willebrand factor and factor VIII can be found (29). Therapeutic trials of heparins, coumadin, and antiplatelet agents have been limited, yielding inconclusive information, but they are ongoing.
Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
HEMOGLOBINS WITH ALTERED OXYGEN AFFINITY
The hemoglobin loading and unloading of oxygen can be expressed by an oxygen dissociation
curve. The physiologic consequences of the abnormal hemoglobins depend on the oxygen affinity,
which defines the point of 50% saturation (p50). The oxygen dissociation curve of normal
hemoglobin represents the reaction of hemoglobin with oxygen as modified by hydrogen ions (Bohr
effect) and 2,3-bisphosphoglycerate (BPG) (30, 31). Hemoglobin oxygen affinity increases with
falling temperature and decreases with rising pH and 2,3-BPG. Hence, red blood cells containing
such an abnormal hemoglobin may have an abnormal oxygen dissociation curve because of 1) an
intrinsic abnormality of hemoglobin-oxygen dissociation, 2) an altered interaction of hemoglobin
with BPG, 3) an altered Bohr effect, or 4) a combination of any or all of the above. It is common to
speak of the oxygen-dissociation curve as being shifted to the left (increased oxygen affinity) or to
the right (decreased oxygen affinity).
POSSIBLE CLINICAL DEVELOPMENTS IN HEMOGLOBIN
Increased oxygen affinity
Some hemoglobins have been described in which the associated clinical manifestations can be
ascribed to an increased oxygen affinity (Table 1). High-affinity hemoglobins bind oxygen more readily and deliver less oxygen to tissues.
Several hemoglobins with increased oxygen affinity have substitutions affecting the α1β2 contact of the tetramer. Others have substitutions involving the C-terminal residues of the beta chain or of the BPG binding sites. All these substitutions favor the oxygenated conformation and cause a left shift of the oxygen dissociation curve, which reflects an increased blood affinity for oxygen. Therefore, it follows that the red cells of such individuals give up less oxygen to the tissues. The relative anoxia increases erythropoietin production and causes polycythemia.
Most of the abnormal hemoglobins with increased oxygen affinity manifest themselves by causing polycythemia in the carrier. The increased oxygen affinity reduces tissue oxygen delivery, causing an increase in erythropoietin secretion and in red cell mass. The possibility of an abnormal hemoglobin with high oxygen affinity should be considered in those atypical patients with polycythemia in which the white blood cell and platelet counts are not elevated and splenomegaly is absent. The importance of establishing the correct diagnosis is mainly to protect the patient from the chemotherapeutic treatment of polycythemia. Family members should be advised that their children may be affected. The life expectancy of affected individuals is essentially normal, and most patients are symptom free. However, if such patients become symptomatic and their hematocrit rises towards 60%, then phlebotomy may be necessary to reduce blood viscosity. Reduced oxygen affinity Only a handful of abnormal hemoglobins have been reported in which a reduced oxygen affinity is the sole abnormality (Table 2). Because of the increased oxygen delivery resulting from the low oxygen affinity, it might be expected that the erythropoietin response would be reduced and these variants would be associated with mild anemia. While this response occurs in most of these variants, it is not so with Hb Kansas carriers. With Hb Kansas, the oxygen affinity is so low that even at normal arterial oxygen tensions there is sufficient desaturation to give rise to clinical cyanosis. The possibility of low-affinity hemoglobins should be considered in patients with low hematocrit or cyanosis with no other cause apparent after evaluation. The p50 is usually elevated. Despite these findings, patients usually require no specific treatment once the correct diagnosis is established. THE UNSTABLE HEMOGLOBINS
At the molecular level, considering the three-dimensional model of the hemoglobin molecule, it
would appear that the stability of the hemoglobin tetramer is dependent on both the internal
molecular positioning of nonpolar amino acids and the stability of the large α1β1 contacts. These
properties serve to hold the four chains together. In most unstable hemoglobins one or more of these
Unstable hemoglobins are hemoglobins that, because of the nature of the substitution, deletion, or
insertion of amino acids (Table 3), tend to undergo spontaneous oxidation within the red cell and
Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
precipitate to form insoluble inclusions called Heinz bodies. Their presence results in the so-called
congenital Heinz body hemolytic anemia. Most patients with this condition are found to have a
nonspherocytic hemolytic anemia. The anemia is exacerbated by infections and oxidative drugs such
as sulfonamides, pyridium, and antimalarials. It must be remembered that the normal red cell is
undergoing continual physical stress and has to be able to deform in arterioles in order to travel
through the microcirculation. The insoluble Heinz bodies are torn out of the red cell during passage
in the microcirculation of the spleen, which is ≤3 microns across (47). In such circumstances, Heinzbodies are pitted out of the red cell along with some membrane, leading to the presence of “bitecells” in the peripheral smear. Other disturbances such as K+ and Ca++ changes are secondary to the physical damage caused by Heinz bodies.
The first report of a child with idiopathic congenital non-spherocytic hemolytic anemia associated with cyanosis and splenomegaly is attributed to Cathie (48). The patient was a small boy. His spleen was removed and, several months later, the red cells were found to contain numerous Heinz bodies. In 1966 Carrell et al described the amino acid substitution giving rise to an unstable hemoglobin (Hb Köln) as the cause of this anemia (42).
The clinical findings in patients suffering from unstable hemoglobin disease include neonatal jaundice, anemia, cyanosis, pigmenturia, splenomegaly, and drug intolerance. The severity of the disease is highly dependent on the degree of instability of the abnormal hemoglobins. The disorder is clearly expressed in heterozygotes, and it seems likely that with most substitutions or deletions, homozygosity would be lethal. Heinz bodies are usually not seen until the spleen has been removed; they can be detected in the peripheral smear by supravital staining. Unstable hemoglobins are detected by their precipitation in isopropanol or after heating to 50°C. HbA2 and HbF may be increased. Hemoglobin electrophoresis reveals that most unstable hemoglobins migrate like HbA or HbS. Complete characterization includes amino acid sequencing and gene cloning and sequencing.
Not for the first time, observations made on patients suffering from certain abnormal hemoglobin have provided the stimulus for basic scientific work. HEMOGLOBIN M AND METHEMOGLOBINEMIA
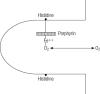
For hemoglobin to combine with oxygen, its iron atoms must be in the ferrous state. Should
oxidation (or de-electronation) of the hemoglobin molecule occur, the ferrous iron is converted to
ferric iron and methemoglobin is formed. Methemoglobin is valueless as a respiratory pigment.
Every day about 1% of the total circulating hemoglobin concentration is converted into
methemoglobin. The iron is itself attached on one side of the “heme pocket” to the amino acid
residue histidine—the proximal histidine. Another histidine is situated on the other side of the
pocket. This second histidine is not directly linked to the ferrous atom and is called the distal
histidine. Normally oxygen is able to slip freely between the distal histidine and the ferrous atom
during oxygenation and deoxygenation (Figure 3). In the normal individual there is a balance
between the spontaneous process of methemoglobin formation and a series of protective
mechanisms that reconvert the pigment back to hemoglobin.
POSSIBLE CLINICAL DEVELOPMENTS IN HEMOGLOBIN
Methemoglobinemia may be caused by the ingestion of nitrites and nitrobenzenes, enzyme
deficiencies such as methemoglobin reductase or diaphorases, and certain abnormal hemoglobins. In
1948 Hörlein and Weber (49) described a German family in which some members had been cyanotic from birth and found that the abnormality was associated with the globin and not with the heme. Hemoglobin M has subsequently been recognized as a perfect example of a molecular abnormality. Such abnormal hemoglobins, collectively called hemoglobin M, all have amino acid substitutions involving either the histidyls themselves or amino acids lining the heme pocket (Table 4). Carriers of hemoglobin M are often cyanotic and suffer from anemia. The anemia is more significant than the hemoglobin level suggests because some 25% of circulating hemoglobin is in the ferric form and therefore is not functional. No effective treatment exists for the cyanosis that is present in patients with hemoglobin M.
Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
POSSIBLE CLINICAL DEVELOPMENTS IN HEMOGLOBIN RESEARCH
It is well known that crocodiles kill their prey by drowning them. Crocodiles are capable of
remaining under water without surfacing to breathe for more than an hour. It has been shown that
when crocodiles hold their breath, bicarbonate ions, the final product of respiration, accumulate and
markedly reduce the oxygen affinity of their hemoglobin. This releases a large fraction of
hemoglobin-bound oxygen to the tissues (55). Hence, the oxygen affinity of crocodile hemoglobin is
markedly reduced by the physiologic concentration of carbon dioxide. The bicarbonate ions thus
formed bind to deoxyhemoglobin and facilitate the giving up of oxygen (Figure 2).
Amino acid sequence identity between crocodile and human hemoglobin is 68% for the alpha
subunit and 51% for the beta subunit. In crocodile hemoglobin, the amino acid residues involved in
bicarbonate ion binding are located at the α1β2 contact. This junction acts as a flexible joint during
In collaboration with Osaka University in Japan, Jeremy Tame at the MRC Laboratory of Molecular Biology in Cambridge, England, was able to transplant this unique allosteric effect from the Nile crocodile (Crocodylus niloticus) into human hemoglobin by replacing a total of 12 amino acids at critical positions in the alpha and beta chains. This new engineered hemoglobin was named Hb Scuba(56). The clinical implication of this work for transfusion medicine is mind-boggling!
Acknowledgment I am deeply grateful to the late Dr. Max Perutz and Professor Herman Lehmann, who first stimulated my interest in hemoglobinopathies, and various commanders in the Royal Air Force and Special Air Service for much assistance. Also, I would like to thank Kathy Cypert (née Martin) for her patient and determined secretarial fortitude. References
1. Perutz MF, Rossmann MG, Cullis MG, Muirhead H, Will G, North ACT. Structure of haemoglobin. A
three-dimensional Fourier synthesis at 5.5Å resolution, obtained by X-ray analysis. Nature. 1960;(185):416–422.
2. Ingram VM. Gene mutations in human haemoglobin: the chemical difference between normal and sickle cell
haemoglobin. Nature. 1957;(180):326–328. [PubMed]
3. Allison AC. Protection afforded by sickle-cell trait against subtertian malarial infection.
Br Med J. 1954;(1):290–294. [PubMed]
4. Perutz MF, Lehmann H. Molecular pathology of human haemoglobin. Nature. 1968;219(157):902–909. [PubMed]
5. Marengo-Rowe A. Haemoglobinopathies. Br J Hosp Med. 1971;6:617–630.
6. Perutz, MF. Proteins and Nucleic Acids: Structure and Function. Amsterdam: Elsevier; 1962. pp. 35–48.
7. Konotey-Ahulu FID. Hereditary qualitative and quantitative erythrocyte defects in Ghana. An historical and
geographical survey. Ghana Med J. 1968;(7):118–119.
8. Herrick JB. Peculiar elongated and sickle-shaped red corpuscles in a case of severe anemia.
9. Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337(11):762–769. [PubMed]
10. Raphael RI. Pathophysiology and treatment of sickle cell disease. Clin Adv Hematol Oncol. 2005;3(6):492–505.
11. Pauling L, Itano HA, Singer SJ, Wells IC. Sickle cell anemia, a molecular disease. Science. 1949;(110):543–548.
12. Nagel, RL.; Platt, OS. General pathophysiology of sickle cell anemia. In: Steinberg MH, Forget BG, Higgs DR. ,
editors. Disorders of Hemoglobin. Cambridge: Cambridge University Press; 2001. pp. 494–526.
13. Pathare A, Kindi SA, Daar S, Dennison D. Cytokines in sickle cell disease. Hematology. 2003;8(5):329–337.
14. Hebbel, RP.; Mohandas, N. Cell adhesion and microrheology in sickle-cell disease. In: Steinberg MH, Forget BG,
Higgs DR. , editors. Disorders of Hemoglobin. Cambridge: Cambridge University Press; 2001. pp. 527–549.
15. Adams RJ, Brambilla D, Optimizing Primary Stroke Prevention in Sickle Cell Anemia (STOP 2) Trial Investigators.
Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease. N Engl J Med. 2005;353(26):2769–2778. [PubMed]
16. Marengo-Rowe AJ, Beale D, Lehmann H. New human haemoglobin variant from southern Arabia: G-Audhali
(alpha-23B4 glutamic acid→valine) and the variability of B4 in human haemoglobin. Nature. 1968;219(159):1164–1166. [PubMed]
Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
17. Perutz RR, Ligouri AM, Eirich F. X-ray and solubility studies of the haemoglobin of sickle-cell anaemia patients.
Nature. 1951;167(4258):929–931. [PubMed]
18. Ballas SK, Smith ED. Red blood cell changes during the evolution of the sickle cell painful crisis.
Blood. 1992;79(8):2154–2163. [PubMed]
19. Vichinsky EP. Comprehensive care in sickle cell disease: its impact on morbidity and mortality.
Semin Hematol. 1991;28(3):220–226. [PubMed]
20. National Heart, Lung, and Blood Institute, National Institutes of Health. The Management of Sickle Cell Disease (NIH Publication No. 02-2117). Bethesda, MD: NIH, 2002. Available at http://www.nhlbi.nih.gov/health/prof/blood/sickle/; accessed February 13, 2006.
21. Vermylen C, Cornu G. Hematopoietic stem cell transplantation for sickle cell anemia.
Curr Opin Hematol. 1997;4(6):377–380. [PubMed]
22. Steinberg MH, Barton F, Castro O, Pegelow CH, Ballas SK, Kutlar A, Orringer E, Bellevue R, Olivieri N, Eckman
J, Varma M, Ramirez G, Adler B, Smith W, Carlos T, Ataga K, DeCastro L, Bigelow C, Saunthararajah Y, Telfer M, Vichinsky E, Claster S, Shurin S, Bridges K, Waclawiw M, Bonds D, Terrin M. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA. 2003;289(13):1645–1651. [PubMed]
23. Cokic VP, Smith RD, Beleslin-Cokic BB, Njoroge JM, Miller JL, Gladwin MT, Schechter AN. Hydroxyurea
induces fetal hemoglobin by the nitric oxide–dependent activation of soluble guanylyl cyclase. J Clin Invest. 2003;111(2):231–239. [PubMed]
24. Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP. Mortality in sickle cell disease.
Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639–1644. [PubMed]
25. Udani M, Zen Q, Cottman M, Leonard N, Jefferson S, Daymont C, Truskey G. Basal cell adhesion
molecule/Lutheran protein. The receptor critical for sickle cell adhesion to laminin. J Clin Invest. 1998;101(11):2550–2558. [PubMed]
26. Ataga KI, Orringer EP. Hypercoagulability in sickle cell disease: a curious paradox.
Am J Med. 2003;115(9):721–728. [PubMed]
27. Westwick J, Watson-Williams EJ, Krishnamurthi S, Marks G, Ellis V, Scully MF, White JM, Kakkar VV. Platelet
activation during steady state sickle cell disease. J Med. 1983;14(1):17–36. [PubMed]
28. Westerman MP, Green D, Gilman-Sachs A, Beaman K, Freels S, Boggio L, Allen S, Zuckerman L, Schlegel R,
Williamson P. Antiphospholipid antibodies, proteins C and S, and coagulation changes in sickle cell disease. J Lab Clin Med. 1999;134(4):352–362. [PubMed]
29. Francis RB Jr. Platelets, coagulation, and fibrinolysis in sickle cell disease: their possible role in vascular occlusion.
Blood Coagul Fibrinolysis. 1991;2(2):341–353. [PubMed]
30. Perutz MF. Stereochemistry of cooperative effects in haemoglobin. Nature. 1970;228(5273):726–739. [PubMed]31. Perutz MF, Wilkinson AJ, Paoli M, Dodson GG. The stereochemical mechanism of the cooperative effects in
hemoglobin revisited. Annu Rev Biophys Biomol Struct. 1998;(27):1–34. [PubMed]
32. Charache S, Weatherall DJ, Clegg JB. Polycythemia associated with a hemoglobinopathy.
J Clin Invest. 1966;45(6):813–822. [PubMed]
33. Botha MC, Beale D, Isaacs WA, Lehmann H. Hemoglobin J Cape Town-alpha-2 92 arginine replaced by glutamine
beta-2. Nature. 1966;212(64):792–795. [PubMed]
34. Jones RT, Osgood EE, Brimhall B, Koler RD. Hemoglobin Yakina. I. Clinical and biochemical studies.
J Clin Invest. 1967;46(11):1840–1847. [PubMed]
35. Lokich JJ, Moloney WC, Bunn HF, Bruckheimer SM, Ranney HM. Hemoglobin Brigham (α A 100
Hemoglobin variant associated with familial erythrocytosis. J Clin Invest. 1973;52(8):2060–2067. [PubMed]
36. Adamson JW, Parer JT, Stamatoyannopoulos G. Erythrocytosis associated with hemoglobin Rainier: oxygen
equilibria and marrow regulation. J Clin Invest. 1969;48(8):1376–1386. [PubMed]
37. Bunn HF, Bradley TB, Davis WE, Drysdale JW, Burke JF, Beck WS, Layer MB. Structural and functional studies
), a variant associated with compensatory erythrocytosis.
J Clin Invest. 1972;51(9):2299–2309. [PubMed]
38. Jensen M, Oski FA, Nathan DG, Bunn HF. Hemoglobin Syracuse A (α A 143
high-affinity variant detected by special electrophoretic methods. Observations on the auto-oxidation of normal andvariant hemoglobins. J Clin Invest. 1975;55(3):469–477. [PubMed]
39. Reissmann KR, Ruth WE, Nomura T. A human hemoglobin with lowered oxygen affinity and impaired heme-heme
interactions. J Clin Invest. 1961;(40):1826–1833. [PubMed]
40. Imamura T, Fujita S, Ohta Y, Hanada M, Yanase T. Hemoglobin Yoshizuka (G10(108)β asparagine→aspartic
acid): a new variant with a reduced oxygen affinity from a Japanese family. J Clin Invest. 1969;48(12):2341–2348. [PubMed]
41. Imai K, Morimoto H, Kotani M, Shibata S, Miyaji T. Studies on the function of abnormal hemoglobins. II. Oxygen
equilibrium of abnormal hemoglobins: Shimonoseki, Ube II, Hikari, Gifu, and Agenogi. Biochim Biophys Acta. 1970;200(2):197–202. [PubMed]
42. Carrell RW, Lehmann H, Hutchinson HE. Hemoglobin Koln (β-98 va-line→methionine): an unstable protein
causing inclusion-body anaemia. Nature. 1966;210(39):915–916. [PubMed]
43. Dacie JV, Shinton NK, Gaffney PJ Jr, Lehmann H. Haemoglobin Hammersmith (beta-42 (CDI) Phe replaced by
Ser). Nature. 1967;216(5116):663–665. [PubMed]
44. Sakuragawa M, Ohba Y, Miyaji T, Yamamoto K, Miwa S. A Japanese boy with hemolytic anemia due to an
unstable hemoglobin (Hb Bristol). Nippon Ketsueki Gakkai Zasshi. 1984;47(4):896–902. [PubMed]
45. Murari J, Smith LL, Wilson JB, Schneider RG, Huisman TH. Some properties of hemoglobin Gun Hill.
Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
Hemoglobin. 1977;1(3):267–282. [PubMed]
46. Plaseska D, Dimovski AJ, Wilson JB, Webber BB, Hume HA, Huisman TH. Hemoglobin Montreal: a new variant
with an extended beta chain due to a deletion of Asp, Gly, Leu at positions 73, 74, and 75, and an insertion of Ala, Arg, Cys, Gln at the same location. Blood. 1991;77(1):178–181. [PubMed]
47. Winterbourn CC, Carrell RW. Studies of hemoglobin denaturation and Heinz body formation in the unstable
hemoglobins. J Clin Invest. 1974;54(3):678–689. [PubMed]
48. Cathie IAB. Apparent idiopathic Heinz body anaemia. Great Ormond Street J. 1952;(2):43–48. 49. Hörlein H, Weber G. Über Chronishce Familiare Metthämoglobinamie und Eine Modificazation des
Methämoglobins. Dtsch Med Wochenschr. 1948;73:476.
50. Gerald PS, Efron ML. Chemical studies of several varieties of Hb, M.
Proc Natl Acad Sci U S A. 1961;(47):1758–1767. [PubMed]
51. Stavem P, Stromme J, Lorkin PA, Lehmann H. Haemoglobin M Saskatoon with slight constant haemolysis,
markedly increased by sulphonamides. Scand J Haematol. 1972;9(6):566–571. [PubMed]
52. Hayashi N, Motokawa Y, Kikuchi G. Studies on relationships between structure and function of hemoglobin
M-Iwate. J Biol Chem. 1966;241(1):79–84. [PubMed]
53. Hutt PJ, Pisciotta AV, Fairbanks VF, Thibodeau SN, Green MM. DNA sequence analysis proves Hb
M-Milwaukee-2 is due to beta-globin gene codon 92 (CAC→TAC), the presumed mutation of Hb M-Hyde Parkand Hb M-Akita. Hemoglobin. 1998;22(1):1–10. [PubMed]
54. Ham RD, Chitayat D, Cooper R, Bandler E, Eng B, Chui DH, Waye JS, Freedman MH. Hb FM-Fort Ripley:
confirmation of autosomal dominant inheritance and diagnosis by PCR and direct nucleotide sequencing. Hum Mutat. 1994;3(3):239–242. [PubMed]
55. Bauer C, Forster M, Gros G, Mosca A, Perrella M, Rollema HS, Vogel D. Analysis of bicarbonate binding to
crocodilian hemoglobin. J Biol Chem. 1981;256(16):8429–8435. [PubMed]
56. Komiyama NH, Miyazaki G, Tame J, Nagai K. Transplanting a unique allosteric effect from crocodile into human
haemoglobin. Nature. 1995;373(6511):244–246. [PubMed]
Figures and Tables Figure 1 Model of the hemoglobin molecule. Two identical white (alpha) polypeptide chains and two identical black (beta) polypeptide chains form a complete molecule. The hemes are shown as discs. O2 marks the oxygen binding site. Reprinted courtesy of Dr. Max (more .) Figure 2 Diagrammatic representation of oxygen equilibrium curves of the lug worm, man, and hemoglobin
Scuba. The effect of hydrogen ions, 2, 3-bisphosphoglycerate, and carbon dioxide (H++BPG +CO2) is to promote a right shift. If man had the hemoglobin of the (more .)
Figure 3 Diagrammatic representation of the heme pocket formed by amino acids. Oxygenation can occur only between the non-heme-linked histidine and iron. Table 1 Examples of hemoglobins with increased oxygen affinity Table 2 Examples of hemoglobins with reduced oxygen affinity
Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
Table 3 Examples of unstable hemoglobins Table 4 Examples of hemoglobin M variants
NIH | Department of Health and Human Services
Privacy Policy | Disclaimer | Freedom of Information Act
Confissões de um ex-cego Sofrimento e Esperança Pr Luciano R. Peterlevitz – Missão Batista Vida Nova, 10/10/2010 Introdução Lemos no Evangelho de João 9 a cura de um cego de nascença. Será que ele já tinha perdido a esperança de cura? No v.25, há uma confissão fantástica: “eu era cego e agora estou enxergando” . São palavras que testemunham o poder de Deus.
SCHEDA DI DATI DI SICUREZZA secondo la Direttiva (EU) No 1907/2006 PULSAR ENERGY BAR 2,6 KG I/GR/F/NL/E/P 000000000000708577 1. IDENTIFICAZIONE DELLA SOSTANZA/PREPARATO E DELLA SOCIETÀ/DELL'IMPRESA Informazioni sul prodotto : PULSAR ENERGY BAR 2,6 KG I/GR/F/NL/E/P Unicamente ad uso di utilizzatori professionali. Via Torino 25 20063 Cernusco sul Naviglio 2. IDENTIFICA






 Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
Note: Performing your original search, "hemoglobin koln" symptoms, in PubMed Central will retrieve 8 citations.
Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
Note: Performing your original search, "hemoglobin koln" symptoms, in PubMed Central will retrieve 8 citations. Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
Hemoglobin has played a spectacular role in the history of biology, chemistry, and medicine. This paper, written primarily for the clinician, is a brief outline of the complex problems associated with abnormal hemoglobins. The thalassemias have been intentionally omitted and will be presented in a separate publication.
Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
Hemoglobin has played a spectacular role in the history of biology, chemistry, and medicine. This paper, written primarily for the clinician, is a brief outline of the complex problems associated with abnormal hemoglobins. The thalassemias have been intentionally omitted and will be presented in a separate publication. Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
The structure of hemoglobin has been extensively studied by x-ray analysis (6). The arrangement ofthe subunits—which is known as the quaternary structure—differs in the oxy- anddeoxyhemoglobin.
Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
The structure of hemoglobin has been extensively studied by x-ray analysis (6). The arrangement ofthe subunits—which is known as the quaternary structure—differs in the oxy- anddeoxyhemoglobin.
 Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
HEMOGLOBINS WITH ALTERED OXYGEN AFFINITY
Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
HEMOGLOBINS WITH ALTERED OXYGEN AFFINITY Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
precipitate to form insoluble inclusions called Heinz bodies. Their presence results in the so-called
congenital Heinz body hemolytic anemia. Most patients with this condition are found to have a
nonspherocytic hemolytic anemia. The anemia is exacerbated by infections and oxidative drugs such
as sulfonamides, pyridium, and antimalarials. It must be remembered that the normal red cell is
undergoing continual physical stress and has to be able to deform in arterioles in order to travel
through the microcirculation. The insoluble Heinz bodies are torn out of the red cell during passage
in the microcirculation of the spleen, which is ≤3 microns across (47). In such circumstances, Heinzbodies are pitted out of the red cell along with some membrane, leading to the presence of “bitecells” in the peripheral smear. Other disturbances such as K+ and Ca++ changes are secondary to the physical damage caused by Heinz bodies.
Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
precipitate to form insoluble inclusions called Heinz bodies. Their presence results in the so-called
congenital Heinz body hemolytic anemia. Most patients with this condition are found to have a
nonspherocytic hemolytic anemia. The anemia is exacerbated by infections and oxidative drugs such
as sulfonamides, pyridium, and antimalarials. It must be remembered that the normal red cell is
undergoing continual physical stress and has to be able to deform in arterioles in order to travel
through the microcirculation. The insoluble Heinz bodies are torn out of the red cell during passage
in the microcirculation of the spleen, which is ≤3 microns across (47). In such circumstances, Heinzbodies are pitted out of the red cell along with some membrane, leading to the presence of “bitecells” in the peripheral smear. Other disturbances such as K+ and Ca++ changes are secondary to the physical damage caused by Heinz bodies.
 Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
POSSIBLE CLINICAL DEVELOPMENTS IN HEMOGLOBIN
Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
POSSIBLE CLINICAL DEVELOPMENTS IN HEMOGLOBIN 

 Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
Hemoglobin. 1977;1(3):267–282. [PubMed]
46. Plaseska D, Dimovski AJ, Wilson JB, Webber BB, Hume HA, Huisman TH. Hemoglobin Montreal: a new variant
with an extended beta chain due to a deletion of Asp, Gly, Leu at positions 73, 74, and 75, and an insertion of Ala, Arg, Cys, Gln at the same location. Blood. 1991;77(1):178–181. [PubMed]
47. Winterbourn CC, Carrell RW. Studies of hemoglobin denaturation and Heinz body formation in the unstable
hemoglobins. J Clin Invest. 1974;54(3):678–689. [PubMed]
48. Cathie IAB. Apparent idiopathic Heinz body anaemia. Great Ormond Street J. 1952;(2):43–48.
Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
Hemoglobin. 1977;1(3):267–282. [PubMed]
46. Plaseska D, Dimovski AJ, Wilson JB, Webber BB, Hume HA, Huisman TH. Hemoglobin Montreal: a new variant
with an extended beta chain due to a deletion of Asp, Gly, Leu at positions 73, 74, and 75, and an insertion of Ala, Arg, Cys, Gln at the same location. Blood. 1991;77(1):178–181. [PubMed]
47. Winterbourn CC, Carrell RW. Studies of hemoglobin denaturation and Heinz body formation in the unstable
hemoglobins. J Clin Invest. 1974;54(3):678–689. [PubMed]
48. Cathie IAB. Apparent idiopathic Heinz body anaemia. Great Ormond Street J. 1952;(2):43–48.
 Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
Table 3
Structure-function relations of human hemoglobins
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1484532
Table 3